What is m5C in RNA?
Among the vast array of RNA modifications that have undergone intensive scrutiny, 5-methylcytosine (m5C) occupies a prominent position as one of the most extensively researched and comprehensively understood. m5C is a prevalent modification found on various RNA molecules, and its presence and role extend across a spectrum of RNA species.
Specifically, m5C plays a pivotal role in diverse cellular processes. It adorns transfer RNAs (tRNAs), where it acts as a regulatory element, exerting control over the translation process. Additionally, m5C is a significant participant in ribosomal ribonucleic acids (rRNAs), where it influences the regulation of ribosomal development and synthesis. Furthermore, m5C modifications grace messenger RNAs (mRNAs), where they exert influence over the structural integrity and stability of mRNA molecules, as well as the intricate orchestration of the translation process itself.
m5C Detection
Recognition of m5C modifications on mRNAs dates back many years, but their comprehensive study was hindered by the scarcity of appropriate tools and methodologies. Technological challenges impeded progress, as conventional techniques lacked the precision and sensitivity required to explore the intricacies of m5C modifications in mRNA molecules.
The resurgence of interest in m5C modifications on mRNAs can be attributed to the development of several cutting-edge research methods, including MeRIP-seq (Methylated RNA Immunoprecipitation Sequencing), miCLIP, Aza-IP, and RNA-BS-seq (RNA Bisulfite Sequencing). These innovative techniques have rekindled the fascination with RNA m5C modifications, placing them back at the forefront of scientific inquiry.
RNA-BS-seq: A Game-Changer for m5C Detection
Among these pioneering methods, RNA-BS-seq is particularly noteworthy for its ability to detect m5C modifications at the single-base resolution level. This technique leverages the principles of bisulfite treatment, which has been widely used in DNA methylation studies. In RNA-BS-seq, RNA is first subjected to bisulfite treatment, which selectively converts unmodified cytosines (C) to uracils (U) while leaving m5C-modified cytosines intact as cytosines. After the bisulfite treatment, PCR is performed to amplify the modified RNA molecules. During PCR, uracils (U) are converted to thymines (T), providing a clear distinction between m5C and unmodified cytosines.

Principle of m5C detection by RNA bisulfite sequencing and combination with next-generation sequencing. (Motorin et al., 2010)
Advantages of RNA-BS-seq for m5C Modification Detection
Targeted mRNA Profiling
RNA-BS-seq is meticulously tailored to specifically target mRNA molecules. This tailored approach allows researchers to streamline their investigations towards comprehending modifications within the transcriptome, rather than encompassing the entire spectrum of RNA species. This precision becomes particularly advantageous when delving into the functional implications of mRNA modifications in gene expression and regulation.
Single-Base Resolution Precision
One of the most salient attributes of RNA-BS-seq is its exceptional capacity to discern m5C modifications at a single-base resolution. This remarkable precision is indispensable for precisely localizing m5C sites within individual mRNA molecules. It empowers researchers not only to pinpoint which cytosines are methylated but also to determine the density and distribution of m5C modifications along the mRNA sequence. Single-base resolution is pivotal for elucidating the potential functional roles of specific modifications within mRNA, encompassing their effects on mRNA structure, stability, and translation efficiency.
Elevated Precision and Reliability
RNA-BS-seq is renowned for its remarkable precision in identifying m5C modifications. Through the utilization of bisulfite treatment and state-of-the-art next-generation sequencing, this technique minimizes the occurrence of false positives and negatives. This rigorous methodology ensures that the identified m5C sites are not only dependable but also biologically significant. The amalgamation of bisulfite conversion and sequencing renders RNA-BS-seq an exceptionally robust and precise tool for profiling m5C modifications throughout the transcriptome.
Workflow of m5C RNA methylation sequencing
I. Total RNA Sample Detection
- Agarose Gel Electrophoresis: This step involves running the RNA samples on an agarose gel. The purpose is to assess the degree of RNA degradation and check for contamination. High-quality RNA will display clear 18S and 28S ribosomal RNA bands on the gel.
- Qubit 2.0 Quantification: The Qubit 2.0 fluorometer is used to accurately quantify the concentration of total RNA in the samples. Typically, an RNA concentration of at least 75 μg is required for downstream processing.
- Agilent 2100 Analysis: It assesses the integrity of the RNA samples. It provides an RNA Integrity Number (RIN) value, with a minimum threshold of 7.5 typically considered acceptable. This step ensures that the RNA molecules are intact and suitable for further processing.
II. Library Construction and Quality Control
- mRNA Extraction: mRNA is extracted from the total RNA using Oligo(dT) magnetic beads. This step selectively captures polyadenylated mRNA molecules, which are the primary targets for m5C sequencing.
- Bisulfite Treatment: The extracted mRNA is subjected to bisulfite treatment. During this process, unmodified cytosines (C) are converted to uracils (U) by bisulfite, while methylated cytosines (m5C) remain as cytosines (C). Zymo-SpinTM IC Columns in the EZ RNAMethylationTM kit are used for mRNA purification during this step.
- Reverse Transcription First-Strand Synthesis: The bisulfite-treated mRNA is reverse transcribed into cDNA. This involves the addition of N6 primer, followed by a series of steps to synthesize the first strand of cDNA from the modified mRNA template.
- Synthesis of a Second Strand of Reverse Transcription: The second strand of cDNA is synthesized by adding second strand buffer, dNTPs, RNase H, and DNA Pol I. This step is carried out in a Thermomixer at a specific temperature. After the reaction, magnetic beads are used for purification, and the eluted product is ready for further processing.
- PCR Amplification: The eluted cDNA is subjected to PCR amplification, which includes end repair, addition of A overhangs, and junction ligation. This amplification step generates the final library of cDNA molecules suitable for sequencing.
III. Library Quality Control
- Initial Quantification: The libraries undergo initial quantification using a Qubit 2.0 fluorometer, with subsequent concentration adjustment to 1 ng/µL as necessary.
- Insert Size Analysis: The size distribution of the libraries is meticulously examined using an Agilent 2100 Bioanalyzer to ensure conformity with expected insert size parameters.
- Accurate Quantification: Precise quantification of library effective concentrations is accomplished through qPCR (quantitative polymerase chain reaction). Libraries are required to possess an effective concentration exceeding 2 nM to meet our rigorous quality standards.
IV. Sequencing
After successfully passing the quality control checks, the libraries are primed for sequencing. Multiple libraries can be strategically pooled based on their effective concentration and the desired volume of sequencing data. Sequencing is typically executed on a platform such as HiSeq, employing the PE150 (paired-end 150) sequencing strategy. This sequencing technique, known as Sequencing by Synthesis, entails the introduction of fluorescently labeled nucleotides, DNA polymerase, and junction primers into the flow cell, where DNA fragments are amplified. The sequencer captures and transforms the fluorescence signals into sequencing data using specialized computer software, thereby furnishing comprehensive sequence information for the segments earmarked for sequencing.
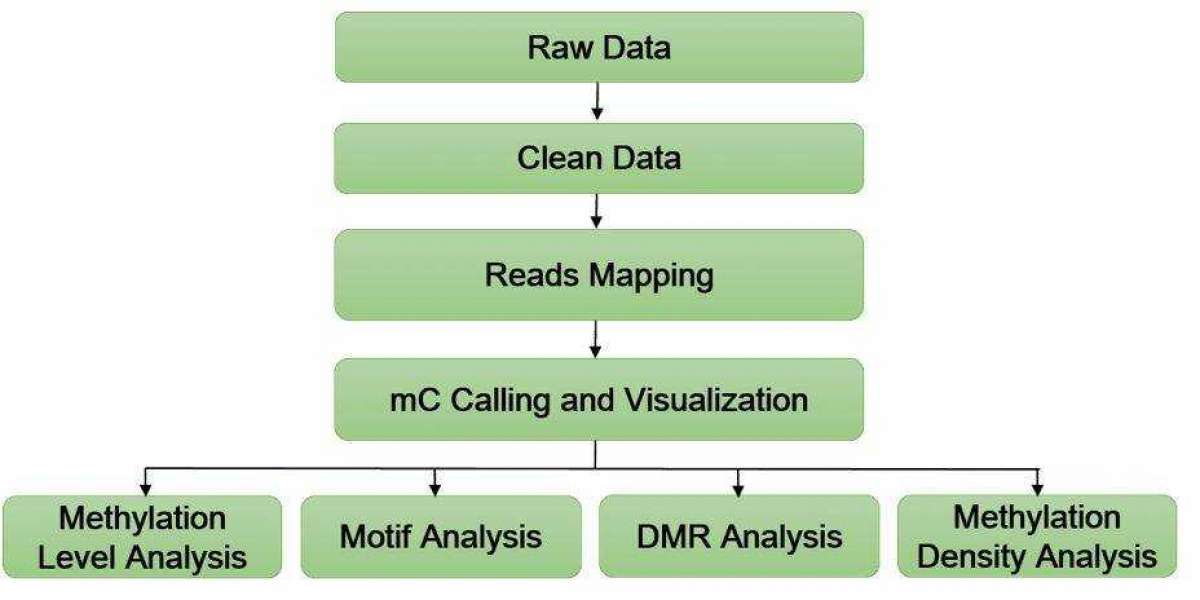
Data Analysis in m5C RNA Methylation Sequencing (RNA-BS)
Data analysis is a critical component of m5C RNA methylation sequencing (RNA-BS) that involves several key steps to ensure the accuracy and reliability of the results. Below, we describe the quality control steps and contrasted quality assessment involved in the data analysis process.

RNA bisulfite sequencing bioinformatics analysis – CD Genomics
Quality Control of Sequencing Data
- Excision of Adapter Sequences: The raw downstream data from sequencing often contains adapter sequences that were introduced during library construction. These adapters need to be removed to ensure that they do not interfere with downstream analysis.
- Average Quality Filtering: Reads with low-quality bases can lead to inaccurate results and reduced data quality. To address this, any read with an average quality score of less than 15 is removed entirely from the dataset.
- Trimming Low-Quality Bases: In addition to filtering low-quality reads, the analysis also involves trimming bases with an average base quality score of less than 3 at the beginning and end of the sequencing reads. This step helps eliminate unreliable sequence information.
- Sequence Length Filtering: Sequences shorter than 20 base pairs (bp) are removed from the dataset after the previous cleanup steps. This filtering is performed to focus on high-quality, informative reads.
Data Quality Assessment
After the initial raw data filtering and preprocessing, data quality assessment is performed to summarize the characteristics of the sequencing data. This assessment typically includes the following checks:
- Sequencing Error Rate Checking: The analysis evaluates the error rate in the sequencing data. High error rates can indicate potential issues with data quality or sequencing chemistry.
- GC Content Distribution Checking: GC content refers to the percentage of guanine (G) and cytosine (C) bases in a sequence. Checking the GC content distribution helps identify any biases in the sequencing process, as imbalanced GC content can affect data quality and mapping efficiency.
Contrasted Quality Assessment
The BS-RNA analysis process is divided into three main steps: preprocessing, contrasting, and annotation. In the contrasting step, various transformations are applied to the reference genome sequences, sequencing data, and gene annotation files.
- Reference Genome Sequence Transformation: The reference genome sequence is modified by replacing cytosine (C) with thymine (T) and guanine (G) with adenine (A). This transformation is done in parallel twice, creating two versions of the reference genome sequence: one with C-to-T changes and the other with G-to-A changes. This transformation facilitates the identification of m5C modifications.
- Transformation of Reads: Reads in the dataset are subjected to transformations based on their nucleotide composition. T-rich reads have their cytosines replaced by thymines (C-to-T transformation), while A-rich reads have their guanines replaced by adenines (G-to-A transformation).
- Mapping with HISAT2: The BS-RNA analysis tool invokes the HISAT2 program to perform mapping and construct alternative splices based on the modified annotated gene files. This mapping process pairs the preprocessed reads with the transformed reference genomic sequence.
After the mapping step, BS-RNA provides several files for further analysis, including:
- Original mapping file in SAM format
- Filtered mapping file in SAM format
- Mapping report file
These files are essential for downstream analysis, annotation, and the identification of m5C modifications within the RNA sequence. The contrasting quality assessment ensures that the analysis is performed accurately and that the modifications are correctly identified and characterized.